

抗感染药物作为医药市场的高频应用品类,其应用范畴已从常规临床治疗延伸至集约化畜禽养殖与水产养殖产业[1-3]。磺胺类、氟喹诺酮类及四环素类等抗生素活性成分,不仅可在亚治疗浓度条件下诱导病原微生物产生耐药性表型,还可通过水平基因转移机制介导耐药基因元件的传播,最终经由食物链与水循环途径进入人体共生菌群,对生态环境稳态及公共健康安全构成双重威胁[4]。据此,构建具备超痕量检测能力、良好精密度及抗干扰性能的分析技术体系,对实现水体中抗生素污染物的有效监测具有重要意义。
超高效液相色谱-串联质谱(UPLC-MS/MS)联用技术凭借其优异的灵敏度、特异性选择性能及复杂基质抗干扰特性,已成为环境水体中抗生素污染物定量分析的首选技术手段。该技术依托色谱分离与质谱检测的协同作用,在痕量有机污染物检测领域优势明显,尤其适用于多组分抗生素残留的精准定性与定量分析。宋焕杰等[5]建立固相萃取(SPE)-UPLC-MS/MS联用方法,实现对水环境中4类15种抗菌药物及其代谢产物的同步测定。张金等[6]采用SPE结合UPLC-MS/MS技术,建立水环境中4种四环素类与6种磺胺类抗生素的快速提取与测定方法,该方法具有操作简便、定性定量准确、检出限低等特点,可满足各类水环境中四环素类与磺胺类抗生素痕量残留的检测需求。
SPE技术是目前文献报道中应用最为广泛的样品前处理方法,在不同研究场景中呈现出多样化的应用模式[7]。自动化SPE系统已成功应用于饮用水源中13类抗生素的痕量富集检测,但该系统存在设备购置成本高昂、运维流程复杂及技术普及门槛较高等不足,限制了其规模化推广应用[8]。常规手动SPE操作多采用3或6 mL标准容量萃取柱,虽可实现500 mL大体积水样的上样处理,但存在操作步骤烦琐、耗时较长及过程控制精度不足等局限性。
鉴于此,本研究采用UPLC-MS/MS分析技术,通过优化SPE自动上样装置,建立水环境中3类24种抗菌药物及其代谢产物的同步测定方法,为水环境中抗生素残留的精准识别与定量分析提供参考。
1 材料与方法
1.1 仪器与试剂
检测仪器为沃特世科技UPLC-TQS 超高压液相色谱-串联三重四极杆质谱仪,配套电喷雾离子化源(ESI+);称量设备选用德国赛多利斯电子天平;样品前处理设备包括Supelco固相萃取装置与Organomation氮吹浓缩仪;固相萃取介质采用沃特世Oasis HLB固相萃取小柱,辅以基本型圆周振荡混合器。
试验所用24种抗菌药物标准物质及对应同位素内标均购自北京曼哈格生物科技有限公司(BePure);色谱级甲醇、乙腈采购于默克公司;色谱级甲酸购自阿拉丁试剂(上海)公司。试验用水由默克Milli-Q Advantage A10超纯水系统制备;过滤耗材为上海新亚0.45 μm水系滤膜与颇尔公司0.22 μm有机系滤膜。
24种抗菌药物标准储备液。(1)磺胺类(12种),磺胺甲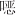 唑(SMX)、磺胺二甲基嘧啶(SMZ)、磺胺间甲氧嘧啶(SMM)、磺胺喹啉(SQ)、磺胺间二甲氧嘧啶(SDM)、磺胺嘧啶(SDZ)、磺胺噻唑(STZ)、磺胺甲基嘧啶(SMR)、磺胺二甲异唑(SIZ)、磺胺甲噻二唑(SMTZ)、磺胺氯哒嗪(SCP)和磺胺多辛(SDX)。(2)氟喹诺酮类(8种),恩诺沙星(ENR)、环丙沙星(CIP)、氧氟沙星(OFX)、洛美沙星(LOM)、达氟沙星(DAN)、沙拉沙星(SAR)、培氟沙星(PEF)和诺氟沙星(NOR)。(3)酰胺醇类(4种),氯霉素(CAP)、甲砜霉素(TAP)、氟苯尼考(FF)和氟苯尼考胺(FFA)。准确移取各目标物标准品溶液,以甲醇定容至10 mL容量瓶,配制成1 μg/mL浓度的混合标准储备液。
唑(SMX)、磺胺二甲基嘧啶(SMZ)、磺胺间甲氧嘧啶(SMM)、磺胺喹啉(SQ)、磺胺间二甲氧嘧啶(SDM)、磺胺嘧啶(SDZ)、磺胺噻唑(STZ)、磺胺甲基嘧啶(SMR)、磺胺二甲异唑(SIZ)、磺胺甲噻二唑(SMTZ)、磺胺氯哒嗪(SCP)和磺胺多辛(SDX)。(2)氟喹诺酮类(8种),恩诺沙星(ENR)、环丙沙星(CIP)、氧氟沙星(OFX)、洛美沙星(LOM)、达氟沙星(DAN)、沙拉沙星(SAR)、培氟沙星(PEF)和诺氟沙星(NOR)。(3)酰胺醇类(4种),氯霉素(CAP)、甲砜霉素(TAP)、氟苯尼考(FF)和氟苯尼考胺(FFA)。准确移取各目标物标准品溶液,以甲醇定容至10 mL容量瓶,配制成1 μg/mL浓度的混合标准储备液。
唑(SMX)、磺胺二甲基嘧啶(SMZ)、磺胺间甲氧嘧啶(SMM)、磺胺喹啉(SQ)、磺胺间二甲氧嘧啶(SDM)、磺胺嘧啶(SDZ)、磺胺噻唑(STZ)、磺胺甲基嘧啶(SMR)、磺胺二甲异唑(SIZ)、磺胺甲噻二唑(SMTZ)、磺胺氯哒嗪(SCP)和磺胺多辛(SDX)。(2)氟喹诺酮类(8种),恩诺沙星(ENR)、环丙沙星(CIP)、氧氟沙星(OFX)、洛美沙星(LOM)、达氟沙星(DAN)、沙拉沙星(SAR)、培氟沙星(PEF)和诺氟沙星(NOR)。(3)酰胺醇类(4种),氯霉素(CAP)、甲砜霉素(TAP)、氟苯尼考(FF)和氟苯尼考胺(FFA)。准确移取各目标物标准品溶液,以甲醇定容至10 mL容量瓶,配制成1 μg/mL浓度的混合标准储备液。24种抗菌药物内标储备液缩写后的下标为同位素标记标识,其中D后数字代表氘代原子的数量,¹³C后数字代表碳-13同位素原子的数量,用于标定对应抗菌药物的同位素标记内标物。(1)磺胺类(12种),磺胺甲唑(SMX-D4)、磺胺二甲基嘧啶(SMZ-13C6)、磺胺间甲氧嘧啶(SMM-13C6)、磺胺喹啉(SQ-13C6)、磺胺间二甲氧嘧啶(SDM-D6)、磺胺嘧啶(SDZ-D4)、磺胺噻唑(STZ-D4)、磺胺甲基嘧啶(SMR-13C6)、磺胺二甲异唑(SIZ-13C6)、磺胺甲噻二唑(SMTZ-13C6)、磺胺氯哒嗪(SCP-D3)、磺胺多辛(SDX-D6)。(2)氟喹诺酮类(8种),恩诺沙星(ENR-D5)、环丙沙星(CIP-D8)、氧氟沙星(OFX-D3)、洛美沙星(LOM-D5)、达氟沙星(DAN-D3)、沙拉沙星(SAR-D8)、培氟沙星(PEF-D5)、诺氟沙星(NOR-D5)。(3)酰胺醇类(4种),氯霉素(CAP-D5)、甲砜霉素(TAP-D3)、氟苯尼考(FF-D3)、氟苯尼考胺(FFA-D3)。准确移取各内标标准品溶液,以甲醇定容至10 mL容量瓶,配制成1 μg/mL浓度的同位素内标混合储备液。所有储备液均于-18 ℃避光条件下保存。
唑(SMX-D4)、磺胺二甲基嘧啶(SMZ-13C6)、磺胺间甲氧嘧啶(SMM-13C6)、磺胺喹啉(SQ-13C6)、磺胺间二甲氧嘧啶(SDM-D6)、磺胺嘧啶(SDZ-D4)、磺胺噻唑(STZ-D4)、磺胺甲基嘧啶(SMR-13C6)、磺胺二甲异唑(SIZ-13C6)、磺胺甲噻二唑(SMTZ-13C6)、磺胺氯哒嗪(SCP-D3)、磺胺多辛(SDX-D6)。(2)氟喹诺酮类(8种),恩诺沙星(ENR-D5)、环丙沙星(CIP-D8)、氧氟沙星(OFX-D3)、洛美沙星(LOM-D5)、达氟沙星(DAN-D3)、沙拉沙星(SAR-D8)、培氟沙星(PEF-D5)、诺氟沙星(NOR-D5)。(3)酰胺醇类(4种),氯霉素(CAP-D5)、甲砜霉素(TAP-D3)、氟苯尼考(FF-D3)、氟苯尼考胺(FFA-D3)。准确移取各内标标准品溶液,以甲醇定容至10 mL容量瓶,配制成1 μg/mL浓度的同位素内标混合储备液。所有储备液均于-18 ℃避光条件下保存。1.2 试验方法
1.2.1 分析条件
色谱条件:采用 Waters Acquity BEH-C18反相色谱柱(2.1 mm×100 mm,1.7 μm),进样量为3 μL。色谱柱温恒定为30 ℃;流动相由含0.01%甲酸的水溶液(A相)和色谱级甲醇(B相)组成。采用梯度洗脱程序(表1),流速设定为0.3 mL/min,以确保组分有效分离。
表1 梯度洗脱程序 |
| 时间/min | 流动相A/% | 流动相B/% | 线性 |
|---|---|---|---|
| 0 | 90 | 10 | |
| 0.5 | 90 | 10 | 6 |
| 3 | 10 | 90 | 6 |
| 4 | 10 | 90 | 6 |
| 7 | 90 | 10 | 1 |
质谱条件:配置电喷雾离子源(ESI),选择正离子扫描模式(ESI+)。离子化参数设置为毛细管电压1.5 kV,去溶剂气温度400 ℃,去溶剂气流速800 L/Hr,以优化分子离子化效率。
1.2.2 样品前处理
取500 mL原始水样经0.45 μm水系滤膜过滤,滤液转移至样品瓶后,精确加入0.1 ng/mL混合内标溶液50 μL。向体系中加入2.92 g磷酸二氢钠与0.25 g Na₂EDTA进行基质调节,并用磷酸溶液将体系pH调至2.34±0.05。固相萃取流程采用Oasis HLB小柱,操作步骤如下:依次用10 mL色谱级甲醇、10 mL超纯水活化固相萃取柱;通过自动化上样系统以5~8 mL/min流速加载上述处理后的水样;用10 mL超纯水淋洗柱床,随后真空干燥20 min;以10 mL甲醇洗脱目标物,洗脱液收集于15 mL离心管中,经氮气吹扫浓缩至近干;加入1 mL含10%甲醇的水溶液复溶,超声混匀后经0.22 μm滤膜过滤,滤液供UPLC-MS/MS分析。
1.2.3 方法线性、检出限和定量限
准确移取24种目标化合物混合标准储备液,经逐级稀释配制浓度为0.5、1.0、2.0、5.0、10.0和20.0 ng/mL的标准工作液,其中内标物浓度恒定为5.0 ng/mL。以各化合物定量离子与对应内标物的质量色谱峰面积比值为纵坐标(y),标准溶液质量浓度为横坐标(x),绘制标准工作曲线并进行线性回归分析,得到各化合物校准方程。方法灵敏度验证依据信噪比(S/N=3)原则,以仪器响应值为基线噪音3倍时对应的标准溶液浓度作为方法检出限(MDLs)。在理论检出浓度附近配制系列加标样品,经系统分析测定,以检出限的3倍作为定量限(MQLs)判定标准。
1.2.4 回收率与精密度
参照前文1.2.2节建立的样品前处理流程,精确移取纯净水样进行阳性加标实验,以评估方法的加标回收率与精密度。分别配制加标浓度为1.0、5.0和10.0 ng/mL的样品,每个浓度水平设置7次平行测定,在相同仪器条件下完成分析,计算各浓度水平的加标回收率及相对标准偏差(RSD)。
2 结果与分析
2.1 分析条件的优化
质谱参数优化在电喷雾正离子模式(ESI⁺)下进行。将24种目标药物单标溶液按优化流速注入质谱仪,启动自动调谐程序,通过一级全扫描模式确定使各化合物分子离子峰强度最大的毛细管电压。以各目标物准分子离子([M+H]⁺)为母离子,进行产物离子扫描,调节碰撞池能量以获得特征碎片离子谱。采用动态多反应监测(MRM)优化策略:选取信噪比(S/N)最高且无同位素干扰的碎片离子作为定量离子,次强且特征性强的碎片离子作为定性离子,组建定性/定量离子对。经系统优化后,确定各化合物的MRM分析条件,具体参数详见表2。
表2 24种化合物及内标物多反应监测条件(MRM) |
| 序号 | 药物 | 母离子/(m/z) | 子离子/ (m/z) | 锥孔电压/V | 碰撞能量/V |
|---|---|---|---|---|---|
| 1 | 磺胺噻唑 | 256.0 | 156.0/108.0 | 20/20 | 15/22 |
| 2 | 磺胺嘧啶 | 251.0 | 156.0/108.0 | 20/20 | 15/25 |
| 3 | 磺胺甲基嘧啶 | 265.0 | 156.0/172.0 | 20/20 | 15/15 |
| 4 | 磺胺二甲基嘧啶 | 279.1 | 186.0/92.0 | 20/20 | 15/30 |
| 5 | 磺胺甲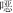 唑 唑 | 254.0 | 92.0/156.0 | 20/20 | 23/12 |
| 6 | 磺胺多辛 | 311.0 | 156.0/108.0 | 20/20 | 20/25 |
| 7 | 磺胺二甲异唑 | 268.0 | 156.0/113.0 | 20/20 | 15/15 |
| 8 | 磺胺喹啉 | 301.1 | 156.0/92.0 | 20/20 | 14/30 |
| 9 | 磺胺间甲氧嘧啶 | 281.1 | 156.0/92.0 | 20/20 | 17/30 |
| 10 | 磺胺间二甲氧嘧啶 | 311.0 | 156.0/92.0 | 20/20 | 18/30 |
| 11 | 磺胺氯哒嗪 | 285.0 | 156.0/108.0 | 20/20 | 15/25 |
| 12 | 磺胺甲噻二唑 | 271.0 | 156.0/108.0 | 20/20 | 15/25 |
| 13 | 环丙沙星 | 332.1 | 288.1/314.1 | 20/20 | 15/15 |
| 14 | 达氟沙星 | 358.2 | 82.2/340.1 | 20/20 | 34/10 |
| 15 | 恩诺沙星 | 360.3 | 316.2/342.1 | 20/20 | 20/20 |
| 16 | 沙拉沙星 | 386.1 | 299.1/342.1 | 20/20 | 28/15 |
| 17 | 培氟沙星 | 334.2 | 233.2/290.4 | 37/37 | 19/24 |
| 18 | 诺氟沙星 | 320.3 | 276.3/302.3 | 10/10 | 16/22 |
| 19 | 氧氟沙星 | 362.3 | 261.2/318.3 | 10/10 | 28/18 |
| 20 | 洛美沙星 | 352.3 | 265.2/308.3 | 10/10 | 25/16 |
| 21 | 氯霉素 | 321.0 | 152.0/257.0 | 30/30 | 20/10 |
| 22 | 甲砜霉素 | 353.9 | 185.1/290.0 | 30/30 | 20/10 |
| 23 | 氟苯尼考 | 355.8 | 119.1/335.9 | 30/30 | 20/10 |
| 24 | 氟苯尼考胺 | 248.1 | 130.0/230.0 | 35/35 | 22/12 |
| 25 | 磺胺噻唑-D4 | 260.0 | 160.0 | 20 | 15 |
| 26 | 磺胺嘧啶-D4 | 255.0 | 160.0 | 20 | 15 |
| 27 | 磺胺甲基嘧啶-13C6 | 271.0 | 162.0 | 20 | 15 |
| 28 | 磺胺二甲基嘧啶-13C6 | 285.2 | 186.0 | 32 | 16 |
| 29 | 磺胺甲唑-D4 | 258.2 | 160.0 | 26 | 16 |
| 30 | 磺胺多辛-D6 | 314.0 | 156.0 | 20 | 18 |
| 31 | 磺胺二甲异-13C6 | 274.0 | 162.0 | 20 | 15 |
| 32 | 磺胺喹啉-13C6 | 307.2 | 162.0 | 28 | 16 |
| 33 | 磺胺间甲氧嘧啶-13C6 | 287.2 | 162.0 | 28 | 18 |
| 34 | 磺胺间二甲氧嘧-D6 | 317.3 | 92.1 | 54 | 32 |
| 35 | 磺胺氯哒嗪-D3 | 314.0 | 156.0 | 20 | 18 |
| 36 | 磺胺甲噻二唑-13C6 | 277.0 | 162.0 | 20 | 15 |
| 37 | 环丙沙星-D8 | 340.2 | 296.2 | 19 | 15 |
| 38 | 达氟沙星-D3 | 361.2 | 343.2 | 20 | 20 |
| 39 | 恩诺沙星-D5 | 365.2 | 245.1 | 20 | 25 |
| 40 | 沙拉沙星-D8 | 384.2 | 303.2 | 20 | 25 |
| 41 | 培氟沙星-D5 | 339.1 | 321.1 | 20 | 18 |
| 42 | 诺氟沙星-D5 | 325.2 | 307.1 | 20 | 18 |
| 43 | 氧氟沙星-D3 | 365.2 | 261.0 | 10 | 25 |
| 44 | 洛美沙星-D5 | 357.2 | 270.2 | 20 | 8 |
| 45 | 氯霉素-D5 | 325.9 | 157.2 | 30 | 18 |
| 46 | 甲砜霉素-D3 | 357.1 | 188.0 | 20 | 20 |
| 47 | 氟苯尼考-D3 | 359.0 | 339.0 | 20 | 5 |
| 48 | 氟苯尼考胺-D3 | 251.2 | 130.0 | 35 | 20 |
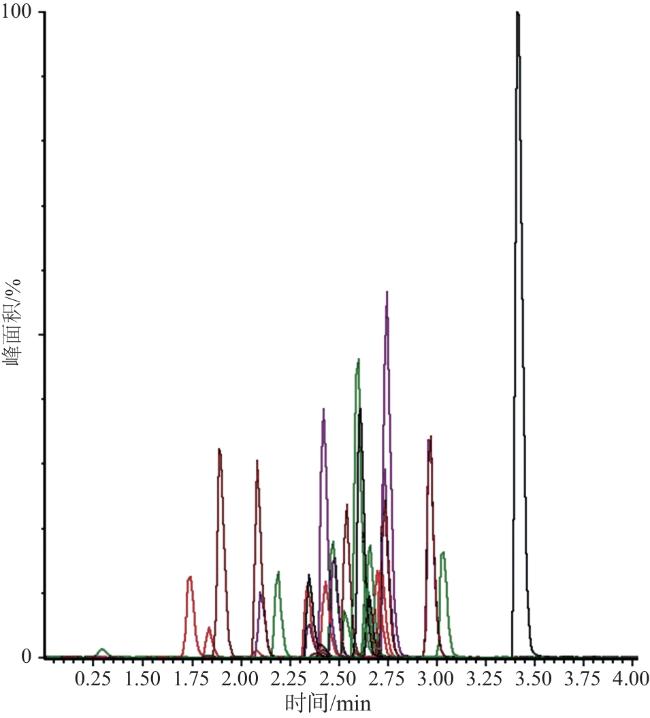
采用0.01%甲酸水-甲醇体系作为流动相,实施梯度洗脱。考察不同进样量对色谱峰形的影响,结果表明,3 μL小体积进样可获得更优峰形,故确定该参数为最终进样量。24种目标药物经UPLC-MS/MS分析所得的MRM色谱如图1所示,各目标物均表现出良好的信号响应及特征碎片离子峰。
{kind=link}
{kind=link}
2.2 方法线性、检出限和定量限
24种化合物的线性相关系数(R²)详见表3,结果显示,其在0.5~20.0 ng/mL浓度范围内均表现出优良的线性响应,R²在0.991 1~0.999 9,表明该方法具备良好的定量线性关系。灵敏度验证结果显示,24种药物的MDLs在0.02~1.00 ng/mL,MQLs在0.06~3.00 ng/mL。该检测体系灵敏度符合相关标准要求,可满足24种药物的日常痕量分析需求。
表3 24种化合物回归方程及相关系数 |
| 序号 | 药物 | 回归方程 | 相关系数(R2 ) | 检出限/(ng/mL) | 定量限/(ng/mL) |
|---|---|---|---|---|---|
| 1 | 磺胺噻唑 | y=0.228 838x-0.047 809 1 | 0.998 082 | 0.12 | 0.36 |
| 2 | 磺胺嘧啶 | y=0.191 854x-0.016 234 8 | 0.996 401 | 0.02 | 0.06 |
| 3 | 磺胺甲基嘧啶 | y=0.225 554x-0.033 521 3 | 0.991 440 | 0.05 | 0.16 |
| 4 | 磺胺二甲基嘧啶 | y=0.236 559x-0.035 562 5 | 0.995 602 | 0.04 | 0.12 |
| 5 | 磺胺甲唑 | y=0.239 951x-0.046 873 1 | 0.994 501 | 0.12 | 0.36 |
| 6 | 磺胺多辛 | y=0.207 532x-0.034 590 9 | 0.996 790 | 0.03 | 0.09 |
| 7 | 磺胺二甲异唑 | y=0.226 76x-0.055 629 5 | 0.995 263 | 0.07 | 0.20 |
| 8 | 磺胺喹啉 | y=0.586 744x-0.044 223 6 | 0.995 963 | 0.05 | 0.14 |
| 9 | 磺胺间甲氧嘧啶 | y=0.293 464x-0.044 914 1 | 0.994 858 | 0.07 | 0.21 |
| 10 | 磺胺间二甲氧嘧啶 | y=0.696 854x-0.053 219 5 | 0.999 899 | 0.30 | 0.90 |
| 11 | 磺胺氯哒嗪 | y=0.215 554x-0.025 586 5 | 0.993 555 | 0.05 | 0.15 |
| 12 | 磺胺甲噻二唑 | y=0.225 654x-0.059 325 8 | 0.994 156 | 0.06 | 0.19 |
| 13 | 环丙沙星 | y=0.402 043x+0.442 194 | 0.994 054 | 0.20 | 0.60 |
| 14 | 达氟沙星 | y=0.076 103 7x+0.421 517 | 0.998 662 | 0.10 | 0.30 |
| 15 | 恩诺沙星 | y=0.437 429x+0.702 566 | 0.998 440 | 0.13 | 0.39 |
| 16 | 沙拉沙星 | y=0.575 35x+1.017 94 | 0.993 139 | 0.15 | 0.45 |
| 17 | 培氟沙星 | y=0.314 479x+0.420 461 | 0.993 164 | 0.29 | 0.87 |
| 18 | 诺氟沙星 | y=0.126 463x+0.118 325 | 0.992 182 | 1.00 | 3.00 |
| 19 | 氧氟沙星 | y=0.611 955x+0.814 736 | 0.991 368 | 0.12 | 0.36 |
| 20 | 洛美沙星 | y=0.278 878x+0.262 32 | 0.993 188 | 0.13 | 0.39 |
| 21 | 氯霉素 | y=0.229 526x+0.010 186 4 | 0.991 107 | 0.05 | 0.15 |
| 22 | 甲砜霉素 | y=0.184 384x+0.010 775 5 | 0.993 387 | 0.26 | 0.78 |
| 23 | 氟苯尼考 | y=0.212 149x+0.007 989 3 | 0.996 321 | 0.20 | 0.60 |
| 24 | 氟苯尼考胺 | y=0.225 414x+0.005 340 48 | 0.995 746 | 0.24 | 0.72 |
2.3 准确度和精密度
各浓度水平的加标回收率及RSD具体数据详见表4。结果显示,24种药物在上述3个加标水平下的回收率在85.9%~113.8%,RSD在1.7%~9.6%,表明本研究建立的分析方法准确度与精密度良好,可满足水体中24种药物残留的日常监测需求。
表4 水体中24种药物的回收率和RSD(n=7) |
| 序 号 | 药物名称 | 添加水平/ (1.0 ng/mL) | 添加水平/ (5.0 ng/mL) | 添加水平/(10.0 ng/mL) | |||
|---|---|---|---|---|---|---|---|
| 回收率 /% | RSD /% | 回收率 /% | RSD /% | 回收率 /% | RSD /% | ||
| 1 | 磺胺噻唑 | 105.1 | 7.3 | 103.9 | 8.9 | 106.5 | 3.5 |
| 2 | 磺胺嘧啶 | 106.1 | 6.6 | 101.1 | 1.7 | 106.6 | 3.5 |
| 3 | 磺胺甲基嘧啶 | 110.9 | 5.4 | 98.1 | 8.1 | 110.8 | 3.3 |
| 4 | 磺胺二甲基嘧啶 | 105.5 | 3.8 | 100.6 | 3.3 | 107.5 | 2.9 |
| 5 | 磺胺甲唑 | 112.0 | 6.1 | 92.3 | 6.9 | 110.5 | 2.7 |
| 6 | 磺胺多辛 | 100.7 | 6.5 | 96.4 | 2.1 | 107.6 | 3.9 |
| 7 | 磺胺二甲异唑 | 109.9 | 4.2 | 96.6 | 2.7 | 107.0 | 5.4 |
| 8 | 磺胺喹啉 | 91.3 | 8.9 | 97.3 | 5.0 | 104.5 | 3.4 |
| 9 | 磺胺间甲氧嘧啶 | 110.9 | 3.8 | 94.3 | 4.1 | 109.4 | 3.9 |
| 10 | 磺胺间二甲氧嘧啶 | 88.1 | 6.3 | 108.0 | 2.2 | 96.0 | 2.9 |
| 11 | 磺胺氯哒嗪 | 107.5 | 8.9 | 100.4 | 2.4 | 109.3 | 4.5 |
| 12 | 磺胺甲噻二唑 | 97.0 | 3.7 | 97.0 | 1.9 | 103.5 | 4.4 |
| 13 | 环丙沙星 | 88.4 | 7.3 | 102.3 | 7.4 | 108.1 | 5.7 |
| 14 | 达氟沙星 | 105.5 | 2.1 | 102.0 | 4.3 | 109.4 | 5.7 |
| 15 | 恩诺沙星 | 87.0 | 8.4 | 100.4 | 3.6 | 106.8 | 3.1 |
| 16 | 沙拉沙星 | 88.0 | 5.4 | 94.6 | 4.6 | 101.1 | 5.2 |
| 17 | 培氟沙星 | 85.9 | 5.5 | 95.7 | 6.8 | 100.9 | 5.5 |
| 18 | 诺氟沙星 | 88.2 | 3.1 | 95.7 | 5.8 | 100.9 | 9.6 |
| 19 | 氧氟沙星 | 107.7 | 5.9 | 101.8 | 4.1 | 108.4 | 3.8 |
| 20 | 洛美沙星 | 87.4 | 6.1 | 92.7 | 6.0 | 97.7 | 7.7 |
| 21 | 氯霉素 | 106.9 | 5.8 | 105.3 | 4.1 | 97.2 | 4.4 |
| 22 | 甲砜霉素 | 91.0 | 2.8 | 98.2 | 3.6 | 96.3 | 6.2 |
| 23 | 氟苯尼考 | 94.2 | 5.6 | 98.1 | 4.6 | 94.3 | 4.1 |
| 24 | 氟苯尼考胺 | 97.8 | 3.4 | 113.8 | 3.8 | 97.4 | 1.9 |
3 结论与讨论
3.1 讨论
3.1.1 固相萃取柱的选择
本研究对2种不同键合相的6 mL规格固相萃取柱(SPE)进行系统评估,分别为反相硅胶型(C₁₈)与亲脂亲水型(HLB)。试验结果表明,在未调节水样pH的条件下,两种SPE柱均能有效富集目标物。对比分析显示,C₁₈柱与HLB柱对24种抗生素及其内标物的回收率均高于75%,但C₁₈柱回收率稳定性欠佳,HLB柱则表现出更优异的回收率与均衡性。基于上述性能差异,本研究最终选用HLB柱作为目标物的净化富集材料。
3.1.2 上样体积的考察
SPE法富集水样的体积主要受SPE柱类型及柱容量限制。本研究选用Oasis HLB柱,通过设置0.5、1.0、1.5 L 3个上样体积梯度,考察目标物回收率以确定最佳上样体积。结果表明,上样量0.5 L时,目标物回收率接近理论最大值,柱容量未达饱和;上样量1.0 L时,回收率略有下降,部分化合物出现穿透现象;上样量1.5 L时,柱体过载导致目标物流失,回收率显著降低。综合回收率与分析效率,最终确定0.5 L为最佳上样体积。
3.1.3 自动上样装置的设计
水体环境中抗菌药物残留检测常采用固相萃取(SPE)柱对目标化合物进行富集净化。针对常规人工上样法处理大体积样品时操作烦琐、耗时久、控制精度低的缺陷,本研究设计了一套自动化上样系统。该系统核心结构由蓄水容器、柔性导流管、液流缓冲腔室、流速调节组件及标准化接口适配器等模块化单元组成。其中,流速调节器通过调控液体流经缓冲腔室的滴落速率实现上样速度动态控制,流速参数经计时器与量具联合校准确定;导流管末端采用密封式接口设计,可适配不同规格SPE柱,保障系统兼容性。将该自动化上样装置与SPE设备集成,构建了大体积水样全流程自动化预处理体系。实验验证表明,该系统兼具操作便捷、运行稳定、成本可控的优势,工程应用价值良好。
3.1.4 液质条件优化
本研究系统考察了液相色谱分离条件对24种目标化合物色谱行为的影响。采用0.01%甲酸甲醇-水体系为流动相,在进样量3 μL、流速0.3 mL/min的梯度洗脱条件下,24种化合物于7 min内实现有效分离,且色谱峰形对称良好。质谱分析采用电喷雾正离子模式(ESI⁺),设定毛细管电压1.5 kV、脱溶剂气温度400 ℃、脱溶剂气流速800 L/h,以多反应监测(MRM)模式定量,结合内标法保障检测数据准确性。方法学验证结果显示,目标物回收率稳定可靠,精密度试验证实仪器分析具备良好重复性。
3.2 结论
(1)亲水亲脂平衡型固相萃取柱(HLB SPE)对目标化合物具有优异保留性能,可简化样品净化流程,实现操作效率与回收效能的双重提升。相较于常规净化方式,该技术无需复杂前处理步骤,在维持高回收率的同时,大幅缩短了样品制备周期。
(2)SPE法富集水样体积主要受柱型及柱容量限制。本研究选用Oasis HLB柱,设置0.5、1.0和1.5 L 3个上样体积梯度,通过考察目标物回收率确定最佳上样量。结果显示,0.5 L上样量时回收率接近理论最大值,柱容量未达饱和;1.0 L时回收率略有下降,部分化合物出现穿透现象;1.5 L时柱体过载导致目标物流失,回收率显著降低。综合回收率与分析效率,最终确定0.5 L为最佳上样体积。
(3)本研究基于超高效液相色谱-串联质谱(UPLC-MS/MS)平台,结合多反应监测(MRM)模式,建立了水体中24种药物的多组分同时定性与定量分析方法。方法学验证结果表明,在1.0、5.0、10.0 ng/mL 3个加标水平下,24种药物平均回收率在85.9%~113.8%,相对标准偏差(RSD)在1.7%~9.6%;方法检出限(MDLs)在0.02~1.00 ng/mL,定量限(MQLs)在0.06~3.00 ng/mL。该方法兼具高灵敏度与良好重复性,适用于大批量水体样品中痕量药物残留的快速筛查与准确定量。