

臭虫隶属于半翅目(Hemiptera)异翅亚目(Heteroptera)臭虫次目(Cimicomorpha)臭虫科(Cimicidae)。该科昆虫无翅,以吸食动物血液为生,常见种类有热带臭虫(Cimex hemipterus)和温带臭虫(Cimex lectularius),被其叮咬后会出现皮肤破损红肿、瘙痒等症状。
昆虫肠道是其生命活动中食物处理与废物排出的核心区域,也是众多微生物的栖息地,其菌落具有复杂性和多样性。昆虫肠道内的微生物群落参与并调节宿主的多种生理机能,包括生长发育、营养吸收与代谢过程,以及对外界环境的适应性[1]。深入解析昆虫肠道细菌群落的结构与功能,对理解昆虫与共生菌间复杂的相互适应机制、协同进化历程及物种分化具有重要意义,且对于开发基于肠道细菌的害虫控制策略具有一定的实践指导意义[2]。常规研究方法受限于可培养细菌的范围,导致大量未知微生物被忽略[3]。16S rRNA高通量测序技术的发展,为全面剖析昆虫肠道菌群结构的复杂性及多样性开辟了新的途径[4]。Upfold等[5]研究指出,大蜡螟(Galleria mellonella)幼虫肠道中定殖的肠球菌可使大蜡螟肠道抗菌肽表达水平上调,对苏云金芽孢杆菌(Bacillus thuringiensis)产生抑制作用。于晓航等[6]通过高通量测序技术分析仁扇舟蛾幼虫肠道不同位置菌群的多样性,为研究肠道细菌在调控仁扇舟蛾与寄主植物互作关系中的作用奠定了基础。王天召等[7]通过高通量测序技术分析了褐飞虱成虫肠道细菌和真菌的多样性,为理解褐飞虱的环境适应性及基于微生物防治技术的开发提供了科学依据。本研究基于16S rRNA序列的高通量测序技术,比较分析了热带臭虫和温带臭虫肠道细菌的多样性,为开发臭虫绿色防控技术提供可利用的微生物资源。
1 材料与方法
1.1 虫源获取及种类鉴定
热带臭虫和温带臭虫于2022年7月采自江苏省南京市栖霞区和燕路某宿舍,带回实验室放置于-80 ℃冰箱保存。于体视显微镜下观察,并根据《热带臭虫及温带臭虫形态与分子鉴定》[8]进行种类鉴定。
1.2 肠道细菌DNA提取
随机挑取热带臭虫(CH)和温带臭虫(CL)各9只,分别分为3组,每组3只。75%乙醇浸泡1 min后,用无菌水漂洗3次。无菌条件下剖取肠道,置于已灭菌的2 mL离心管中,保存于-20 ℃冰箱备用。参照DNA提取试剂盒(DNeasy® Blood &Tissue Kit)的说明书,提取肠道细菌基因组DNA;使用微量分光光度计(Thermo Scientific NANODROP 2000c)检测基因组DNA浓度;用1.2%琼脂糖凝胶电泳检测DNA完整性。检测合格后的样品保存于-80 ℃备用。
1.3 PCR扩增与高通量测序
以提取的肠道细菌总DNA为模板,用引物338F(5'-ACTCCTACGGGAGGCAGCA-3')和806R(5'-GGACTACHVGGGTWTCTAAT-3')对16S rRNA的V3-V4可变区进行PCR扩增[9]。PCR扩增过程:98 ℃预变性2 min;98 ℃变性15 s;55 ℃退火30 s;72 ℃延伸30 s,25~30个循环;72 ℃延伸5 min,4 ℃保存。PCR扩增体系总体积为25 μL:5×reaction buffer 5 μL,5×GC buffer 5 μL,dNTP 2 μL,Forwardprimer 1 μL,Reverseprimer 1 μL,DNA Template 2 μL,ddH2O 8.75 μL,Q5DNAPolymerase 0.25 μL。PCR扩增产物经1.20%琼脂糖凝胶电泳检测,并利用Axygen® AxyPrep DNA Gel Extraction Kit试剂盒进行纯化。检测合格后送至上海派森诺生物科技股份有限公司,采用Illumina NovaSeq PE250测序平台进行双端测序。
1.4 高通量测序数据分析
Illumina NovaSeq测序完成后,利用QIIME2软件的子程序qiime cutadapt trim-paired切除原始双端序列的引物片段,并丢弃未匹配引物的序列。启动qiime dada2 denoise-paired模块,采用DADA2算法将低质量碱基过滤,再将正向与反向reads进行拼接并对序列中的碱基差异进行去噪处理,排除可能因测序错误造成的假阳性变异。最终通过de novo方式检测并剔除PCR反应中产生的嵌合体序列,从而得到样本中高分辨率的扩增子序列变体(ASVs)。
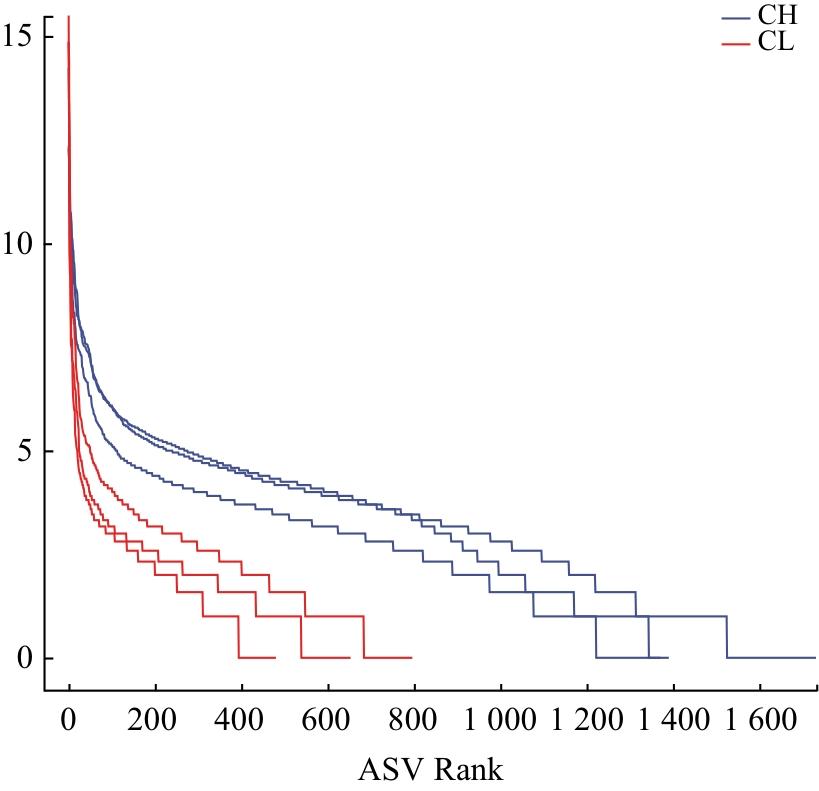
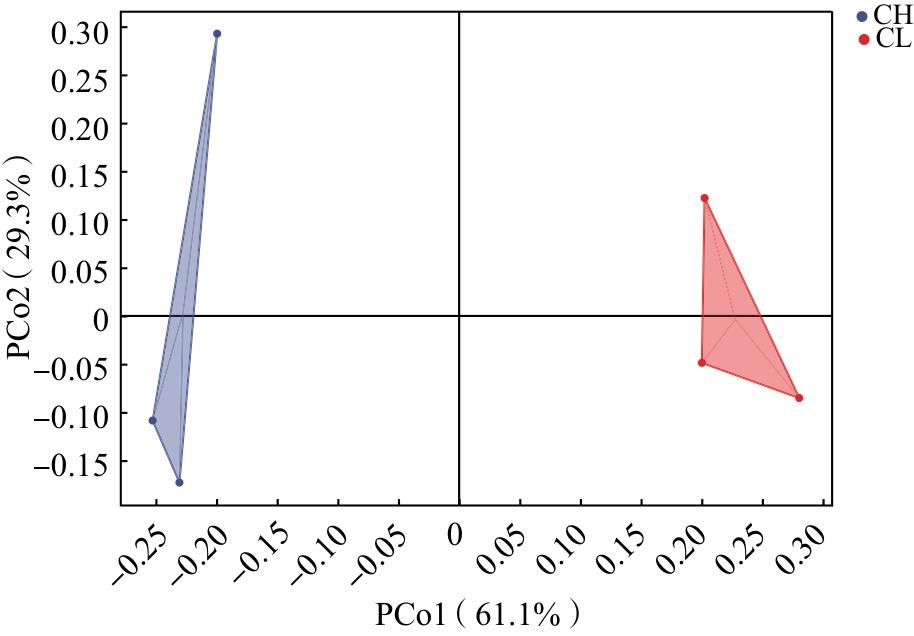
依据Greengenes 13.8数据库中的16S rRNA序列与分类信息建立模型,采用QIIME2的qiime feature-classifier classify-sklearn模块对细菌的16S rRNA的V3-V4区域基因序列进行物种注释。利用R语言计算各样本ASVs的聚类结果,制作物种组成分析图及韦恩图。利用qiime diversity alpha-rarefaction进行α多样性分析,计算α多样性指数,包括观测物种数、Chao1指数、Shannon指数、Simpson多样性指数、Pielou均匀度指数和覆盖度。Rank-abundance曲线可以同时展现样品中所含物种的丰富度和均匀度,横轴长度代表ASVs数,反应物种的丰富度;曲线平缓且逐渐下降,表明样品中细菌菌群的均匀度较高;相反,曲线急剧陡降,则显示优势菌群占据较大比例,从而反映出较低的均匀度。利用QIIME2软件,采用加权UniFrac距离算法的主坐标分析(Principal coordinates analysis,PCoA)评估2种臭虫样本肠道细菌群落组成的差异性(β多样性)。
2 结果与分析
2.1 2种臭虫肠道细菌测序
对热带臭虫和温带臭虫肠道样本的16S rRNA扩增产物进行测序,产生原始序列分别在84 010~145 544,137 165~146 410条,去噪后有效序列分别在75 923~133 067,124 943~132 599条。2种臭虫的肠道细菌通过聚类分析分别获得1 372~1 732,481~797个ASVs(表1)。
表1 热带臭虫和温带臭虫肠道细菌16S rRNA基因测序基本信息 |
| 样品 | 原始序列数 | 有效序列数 | ASVs数 | 不同分类阶元分类单元数量 | |||||
|---|---|---|---|---|---|---|---|---|---|
| 门 | 纲 | 目 | 科 | 属 | |||||
| 热带臭虫 | CHV1 | 84 010 | 75 923 | 1 372 | 30 | 68 | 104 | 138 | 168 |
| CHV2 | 145 544 | 133 067 | 1 391 | 27 | 68 | 95 | 134 | 167 | |
| CHV3 | 143 917 | 128 134 | 1 732 | 27 | 64 | 102 | 154 | 194 | |
| 温带臭虫 | CLV4 | 137 165 | 124 943 | 654 | 19 | 46 | 71 | 108 | 101 |
| CLV5 | 142 865 | 130 460 | 481 | 19 | 48 | 70 | 92 | 88 | |
| CLV6 | 146 410 | 132 599 | 797 | 24 | 56 | 73 | 108 | 102 | |
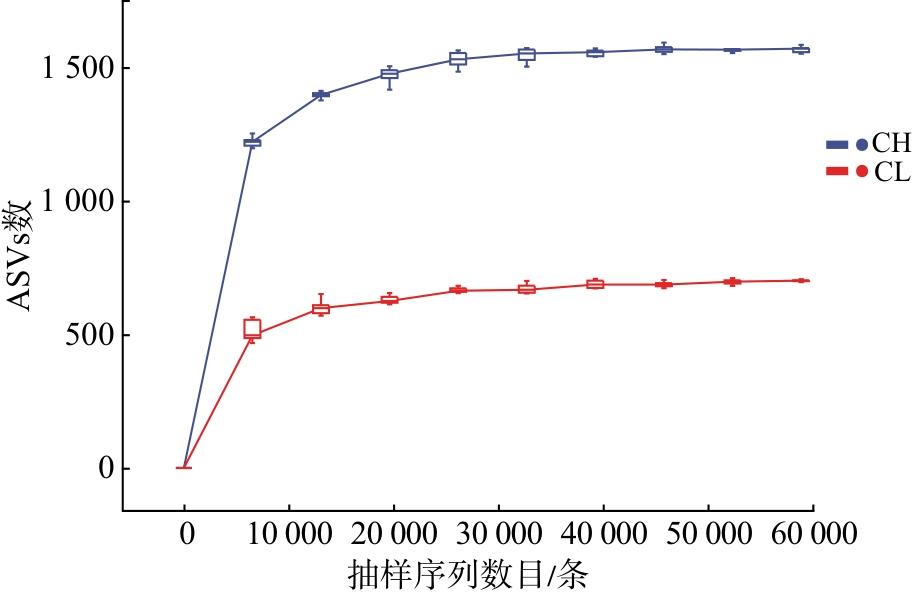
对测序序列进行随机抽样的方法,以抽到的序列数与其代表的ASVs数构建2种臭虫肠道微生物高通量测序结果的稀释曲线。由图1可知,16S rRNA测序的稀释曲线的上升逐渐趋于平缓,说明测序数据量充足。样品中热带臭虫肠道菌群的丰度较温带臭虫的高,且曲线的斜率逐渐降低,表明获得的测序数据量包含热带臭虫和温带臭虫肠道大多数细菌。因此,本研究测序量基本能反映热带臭虫和温带臭虫肠道微生物的组成和种类。
2.2 2种臭虫肠道细菌菌群的韦恩图
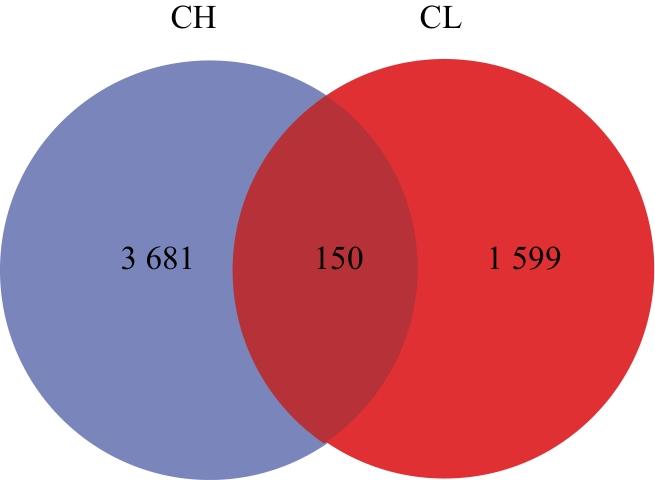
由图2可知,热带臭虫特有的ASV为3 681个,温带臭虫特有的ASV为1 599个,二者共有的ASV数目为150个,热带臭虫肠道细菌群落中可能含有更多独特的物种。
2.3 2种臭虫肠道菌群的群落组成
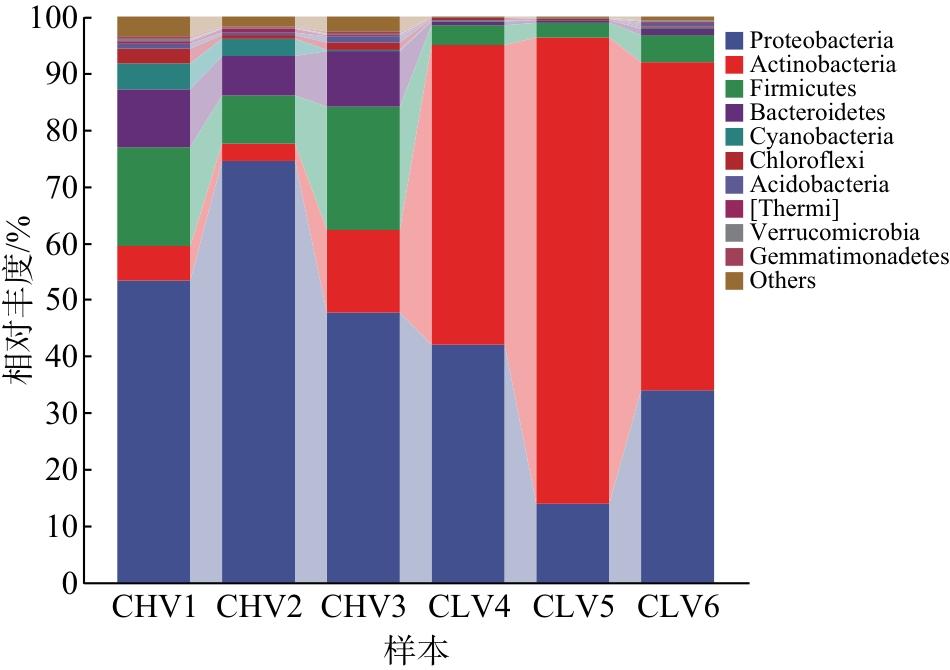
在门分类阶元水平上,热带臭虫和温带臭虫肠道细菌菌群主要由变形菌门(Proteobacteria)、放线菌门(Actinobacteria)和厚壁菌门(Firmicutes)组成(图3)。变形菌门在热带臭虫3个样品中占比分别为53.25%、74.41%、47.66%,在温带臭虫中占比分别为41.94%、14.01%、33.90%。放线菌门在热带臭虫中的丰度较低(6.19%、3.10%、14.44%),在温带臭虫中的丰度较高(53.11%、82.3%、58.08%)。厚壁菌门在热带臭虫中的丰度较高(17.28%、8.51%、21.67%),在温带臭虫中的丰度较低(3.44%、2.51%、4.69%)。此外,热带臭虫肠道菌的优势细菌还有拟杆菌门(Bacteroidetes)和蓝菌门(Cyanobacteria),在热带臭虫3个样品中丰度分别为10.17%、6.84%、9.8%和4.74%、3.06%、0.34%,在温带臭虫中拟杆菌门丰度占比较低(0.63%、0.47%、1.03%),蓝菌门占比更低(0.17%、0.07%、0.17%)。
由图4可知,纲水平上,热带臭虫肠道细菌的优势菌群为α-变形菌纲(Alphaproteobacteria)(27.44%、62.07%、20.39%)、梭菌纲(Clostridia)(12.47%、5.97%、13.08%)、β-变形菌纲(Betaproteobacteria)(12.21%、6.85%、11.47%)、γ-变形菌纲(Gammaproteobacteria)(10.87%、4.15%、13.77%)和腐败螺旋菌纲(Saprospirae)(6.19%、3.42%、5.33%)(图4A)。温带臭虫肠道细菌的优势菌群为放线菌纲(Actinobacteria),丰度为52.90%、82.17%、56.79%;α-变形菌纲的丰度为38.37%、11.63%、17.56%;γ-变形菌纲的丰度为2.45%、1.84%、14.12%,芽孢杆菌纲(Bacilli)的丰度为2.97%、2.35%、3.79%;β-变形菌纲的丰度为1.03%、0.41%、2.06%(图4B)。
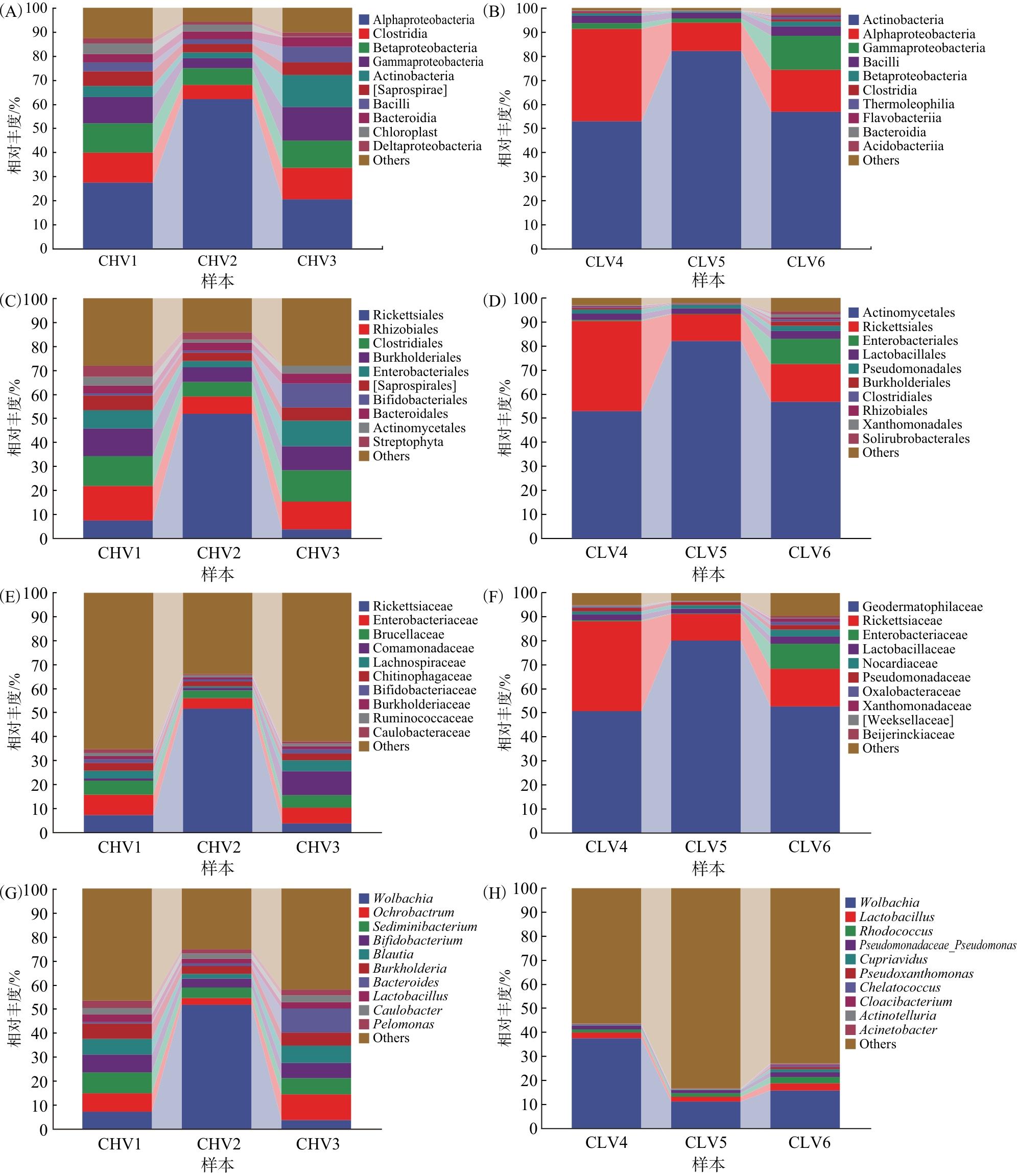
目水平上,热带臭虫肠道细菌的优势菌群为立克次体目(Rickettsiales)(7.42%、51.79%、3.72%)、根瘤菌目(Rhizobiales)(14.33%、7.30%、11.60%)、真杆菌目(Clostridiales)(12.45%、5.90%、13.02%)、伯克氏菌目(Burkholderiales)(11.56%、6.07%、10.03%)和肠杆菌目(Enterobacteriales)(7.57%、2.74%、10.70%)(图4C)。温带臭虫肠道细菌的优势菌群为放线菌目(Actinomycetales)(52.89%、82.14%、56.74%)、立克次体目(37.53%、11.14%、15.84%)和乳杆菌目(Lactobacillales)(2.53%、2.12%、3.28%)。肠杆菌目在温带臭虫中的丰度相差较大,CLV6中丰度为10.47%,而在另2组中丰度占比仅有0.41%和0.22%(图4D)。
科水平上,热带臭虫肠道细菌的优势菌群为立克次体科(Rickettsiaceae)(7.26%、51.70%、3.61%)、肠杆菌科(Enterobacteriaceae)(7.57%、2.74%、10.70%)、布鲁氏菌科(Brucellaceae)(8.57%、4.44%、6.82%)、丛毛单胞菌科(Comamonadacea)(7.54%、3.61%、6.22%)、毛螺菌科(Lachnospiraceae)(6.53%、1.92%、7.32%)和几丁质噬菌科(Chitinophagaceae)(6.19%、3.40%、5.33%)(图4E)。温带臭虫肠道细菌的优势菌群为地嗜皮菌科(Geodermatophilaceae)和立克次体科(37.48%、11.14%、15.77%);在3组样本中地嗜皮菌科占比均超过50%(50.82%、52.70%),最高占比高达80.18%(图4F)。
属水平上,沃尔巴克氏体属(Wolbachia)在热带臭虫和温带臭虫中丰度均最高,在热带臭虫肠道细菌菌群的一个样品占比高达51.7%,其余2个分别为7.27%和3.62%(图4G)。在温带臭虫肠道菌群的3个样品中丰度远高于其他菌群,占比分别为37.49%、11.14%、15.77%。此外,热带臭虫肠道细菌菌群中其他优势属为苍白杆菌属(Ochrobactrum)、沉积杆菌属(Sediminibacterium)和双歧杆菌属(Bifidobacterium),其在3组样品中占比分别为8.46%、4.35%、0.71%,5.92%、3.23%、0.95%,3.26%、2.03%、9.93%。温带臭虫肠道菌群中其他优势属为乳杆菌属(Lactobacillus)、红球菌属(Rhodococcus)和假单胞菌属(Pseudomonas),在3组样品中占比分别为2.29%、1.99%、2.89%,1.38%、1.51%、2.78%,1.48%、1.29%、1.90%(图4H)。在相对丰度较高的前10种菌群中红球菌属为温带臭虫肠道细菌菌群的特有种类。
2.4 2种臭虫肠道细菌的α多样性
表2 热带臭虫和温带臭虫肠道细菌的Alpha多样性指数 |
| 样品 | 观测物种数 | Chao1指数 | Shannon指数 | Simpson多样性指数 | Pielou均匀度 | 覆盖度/% | |
|---|---|---|---|---|---|---|---|
| 热带臭虫 | CHV1 | 1 373 | 1 377.88 | 7.72 | 0.97 | 0.74 | 99 |
| CHV2 | 1 389 | 1 468.54 | 5.53 | 0.86 | 0.53 | 99 | |
| CHV3 | 1 744 | 1 856.72 | 7.95 | 0.98 | 0.73 | 99 | |
| 温带臭虫 | CLV4 | 652 | 712.60 | 2.27 | 0.61 | 0.24 | 99 |
| CLV5 | 487 | 526.30 | 1.58 | 0.36 | 0.17 | 99 | |
| CLV6 | 796 | 863.72 | 3.31 | 0.69 | 0.34 | 99 | |
2.5 2种臭虫肠道细菌群落结构差异
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
3 结论与讨论
在昆虫的所有部位中,肠道的细菌种类最为丰富。本研究基于16S rRNA高通量测序技术,对热带臭虫和温带臭虫的肠道细菌组成和多样性进行分析,揭示了2种臭虫肠道内细菌群落结构的差异性。结果表明,在门分类阶元水平上,热带臭虫和温带臭虫肠道优势菌群均含有变形菌门,在热带臭虫3个样品中,变形菌门占比最高,分别为53.25%、74.41%、47.66%,在温带臭虫3个样品中,变形菌门占比为41.94%、14.01%、33.90%。该结果与鳞翅目昆虫[10]、红火蚁[11]、淡色库蚊[12]等昆虫肠道的优势菌群结果契合。本研究发现,温带臭虫肠道细菌的优势菌群中放线菌门丰度最高,在3个样品中占比分别为53.11%、82.3%和58.08%。表明变形菌门和放线菌门的菌群可能影响着宿主昆虫体内的代谢、生理和行为机制[13-14]。
在属分类阶元上,热带臭虫和温带臭虫肠道共有的优势属为沃尔巴克氏体属。沃尔巴克氏体隶属于变形菌门,是一种普遍存在于节肢动物体内,并对其宿主生殖行为进行调控的共生细菌[15]。Hosokawa等[16]研究证实,沃尔巴克氏体存在于温带臭虫体内,并能提供其生长所需的B族维生素。Lim等[17]对吸血与饥饿状态下的热带臭虫进行对比分析,发现饥饿状态下的臭虫体内细菌菌落较吸血状态下的细菌群落更为丰富。在本研究的热带臭虫的3个样品中,CHV2中沃尔巴克氏体属丰度明显高于其他2个样品,可能是样本采集时CHV1和CHV3正处于吸血后的消化过程,此过程产生的各种消化酶对部分细菌产生抑制作用。而CHV2处于完成消化之后,吸血后的营养供应可能会使肠道内细菌丰富度增加。此外,温带臭虫肠道内沃尔巴克氏体属总体丰度略高于热带臭虫,可能与不同物种的自身特性相关。
热带臭虫肠道细菌菌群中其他优势属为苍白杆菌属、沉积杆菌属和双歧杆菌属。温带臭虫肠道菌群中其他优势属为乳杆菌、红球菌属和假单胞菌属。王天召等[7]在褐飞虱肠道菌群中发现乳杆菌属细菌,认为该菌可能具有合成多糖类化合物降解酶的作用,参与褐飞虱的体内代谢过程。Wang等[18]研究发现,蜂王和工蜂中许多肠道优势细菌菌种具有一致性,但丰度占比存在差异;衰老的蜂王肠道菌群中乳杆菌属和双歧杆菌属细菌的丰度高于衰老的工蜂,推测蜂王能更好地抵御致病菌与这些菌种有关。双歧杆菌属在热带臭虫和温带臭虫肠道中均有存在,可能与营养代谢及免疫作用有关。在舞毒蛾肠道中红球菌属能帮助其降解萜烯类化合物[10],泽兰实蝇肠道内的红球菌属能够降解植物的毒素[19]。假单胞菌属在蚊子[20]等多种昆虫体内发现,其能降解昆虫肠道内的有毒物质。Zhang等[21]研究发现,沉积杆菌属具有降解塑料的能力。这些具有解毒功能的菌属与臭虫抗药性产生的相关性,仍需通过针对性试验进一步探究其具体作用机制。刘婧等[22]对家蝇发育过程中肠道细菌菌群进行研究,发现苍白杆菌属为蝇蛹期和成蝇所共有,推测其可能在从蛹到成蝇的变态过程中起重要作用。此外,在蚜虫、苜蓿盲蝽属、褐飞虱属、瓜蝽属和红猎蝽属等半翅目昆虫肠道中均发现苍白杆菌属菌种,可能参与宿主体内的营养代谢,促进发育[13]。热带臭虫和温带臭虫肠道细菌群落结构差异较大,可能与臭虫的进食情况、肠道内环境以及宿主的遗传特性等因素有关。
综上,肠道菌群在昆虫营养吸收、免疫调节、生长发育及抗病防御等方面发挥关键作用。本研究利用16S rRNA高通量测序技术分析了热带臭虫和温带臭虫肠道菌群的组成和多样性,结果表明,2种臭虫的肠道微生物菌群多样性差异明显,研究结果有利于进一步明确臭虫肠道菌群的生理功能,为开发以菌治虫技术提供依据。