

肠道微生物及其代谢产物在宿主的一系列生物学功能中发挥着重要作用,如营养和能量获取、免疫和炎症调节以及神经传导等[1-2]。寄主系统发育、生长、饮食和健康状况对肠道细菌群落的组成和多样性有一定的影响[3-4]。外部环境中的微生物有助于动物肠道微生物群落的建立。Zhang等[5]研究发现,土壤无脊椎动物的肠道微生物是由土壤微生态带逐渐建立而成。微生物群落分析通常采用分离培养法,但这种方法从样品中分离出微生物有限,因此较难准确地计算出微生物的数量[6]。严琼英等[7]利用聚合酶链式反应-变性梯度凝胶电泳和16S rDNA技术进行菌群结构分析,得出的结果较完整。刘骁蒨等[8]分析了变性梯度凝胶电泳技术在马属动物肠道微生物多样性研究中的应用,表明该技术在分析动物肠道微生物多样性方面具有快速、精确等优点,是筛选优势菌株和分离有害菌的有效手段之一。随着测序技术不断发展,高通量测序成为反映样品中微生物信息的有效工具之一。
1 材料与方法
1.1 样品采集和处理
在黄河三角洲地区进行样品采集。在宽体金线蛭养殖区域设置4块1 m×1 m的样地,清除可见的植物凋落物和石块,从每块样地采集0~10 cm的表层土壤样品,保存于干冰中运回实验室。从土壤中取出宽体金线蛭,放入原始栖息地的土壤中,然后活体带回实验室。随机抽取体重(2.0±0.6)g的宽体金线蛭,放入冰块中,用75%乙醇和无菌蒸馏水清洗。然后在无菌条件下解剖金线蛭标本,收集肠道组织。将采集的肠道组织(GUT)和土壤样品(WS)保存在-80 ℃冰箱中,用于DNA提取。
1.2 高通量测序
使用土壤DNA试剂盒提取土壤样本的总基因组DNA,并使用粪便DNA试剂盒提取水蛭肠道样本的总基因组DNA。使用通用引物341F(5’-CCTACGGGNGGCWGCAG-3’)和805R(5’-GACTACHVGGGGTATCTAATCC-3’)扩增细菌16S rRNA基因的V3-V4区,扩增条件为98 ℃变性30 s;98 ℃变性10 s,54 ℃退火30 s,72 ℃延伸45 s,32个循环;72 ℃延伸10 min[12]。反应混合物包括25 ng模板DNA、12.5 µL预混料、上、下游引物序列各2.5 µL和PCR级水。用2%琼脂糖凝胶电泳法鉴定扩增产物,用AMPure XT纯化,用Qubit定量。在此基础上,建立扩增子库,利用NovaSeq 6000测序仪进行双端2×250 bp测序。
1.3 数据分析
将测序过程、统计分析和双端读数组装成基于条码的样本,切割条码和引物序列。使用FLASH v1.2.8软件合并对端读数。通过筛选嵌合体和去除长度小于100 bp的序列,获得高质量的数据。使用QIIME2软件中的除性扩增去噪算法(DADA2)插件进行复制,得到扩增序列变异(ASV)表,并去除单态ASV,分析细菌多样性组成[13],使用Kruskal-Wallis计算细菌丰富度指数(Chao1指数)和多样性指数(Shannon指数),评价细菌群落丰富度和多样性。利用R语言绘制韦恩图和主坐标分析(Principal coordinates analysis,PCoA)图。进行置换多变量方差分析,对PCoA结果进行统计。使用Python LEfse包,进行线性判别(LEfse)分析,分析组间差异,获得不同分组具有统计学差异的微生物组成。采用PICRUST2对不同分组的菌群开展功能预测分析。
2 结果与分析
2.1 细菌菌群多样性
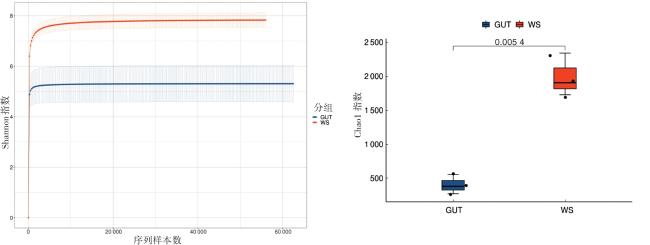
本研究共获得480 044个原始读数。过滤原始读数后,获得479 411个有效读数。由图1可知,肠道组(GUT)和土壤组(WS)中细菌群落的丰富度和多样性存在差异。肠道细菌群落Shannon指数和Chao1指数明显低于土壤组,说明相比于水蛭肠道菌群,土壤细菌群落更加多样化。
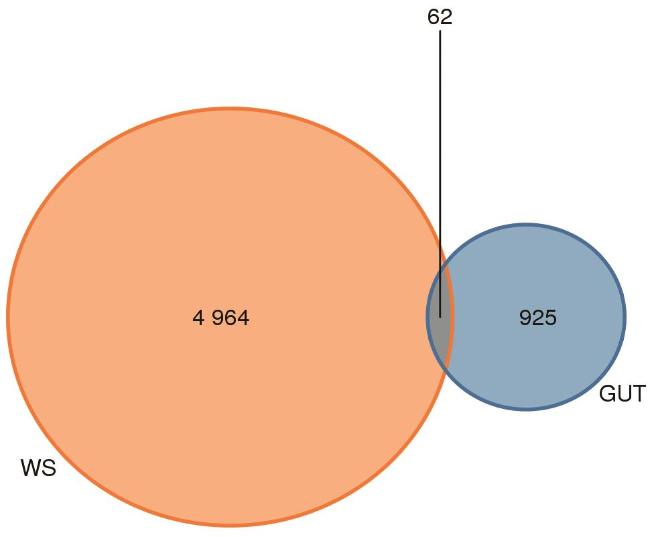
韦恩图(图2)显示了土壤和肠道菌群之间共享和独有的ASV数量。GUT和WS组的ASV数分别为987和5 026个,共有的ASV数量为62个。共享ASV在两组ASV总数中所占比例很小。表明土壤微生物中包含更多的ASV,具有更高的特异性。
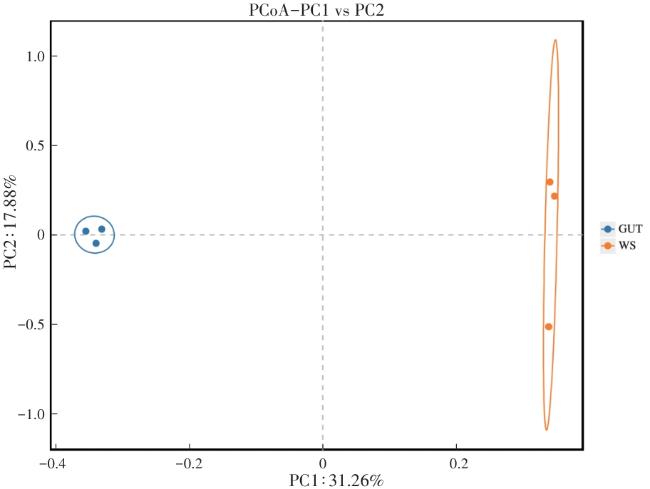
2.2 细菌菌群组成
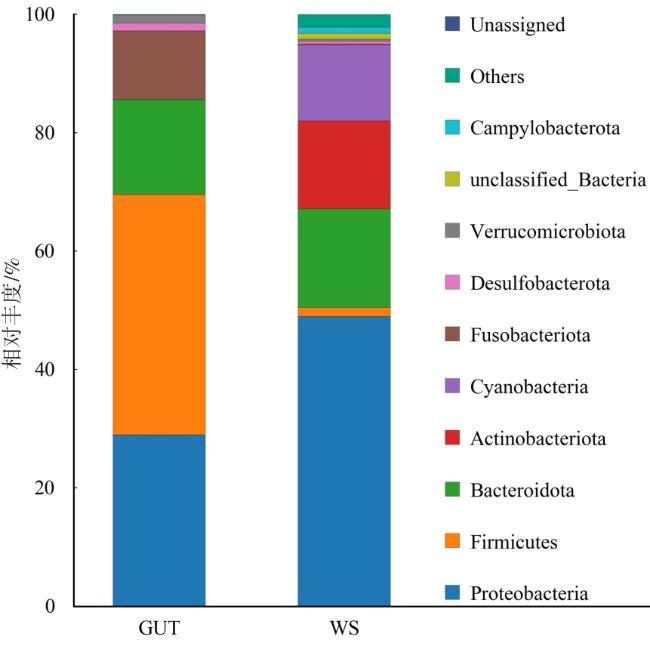
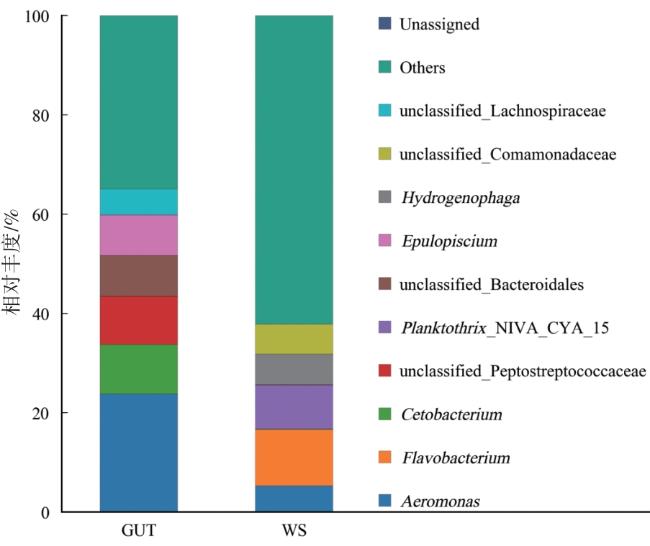
细菌群落在门水平的相对丰度如图4所示,水蛭肠道类群中相对丰度大于1%的类群为厚壁菌门(Firmicutes)40.64%、变形杆菌(Proteobacteria)28.94%、拟杆菌门(Bacteroidota)16.20%和梭杆菌门(Fusobacteriota)11.57%。土壤类群中相对丰度大于1%的类群为变形杆菌48.95%、拟杆菌门16.69%、放线菌门(Actinobacteriota)14.85%、蓝藻菌门(Cyanobacteria)12.87%和厚壁菌门1.52%。变形杆菌是两组菌群中最丰富的类群。
由图5可知,在属水平上,除未分类属外,肠道类群中优势属(相对丰度>1%)为气单胞菌属(Aeromonas)23.76%、鲸杆菌属(Cetobacterium)9.98%和刺骨鱼属(Epulopiscium)8.16%。土壤类群中优势属为氢噬胞菌属(Hydrogenophaga)6.16%、黄杆菌属(Flavobacterium)11.36%和气单胞菌属5.26%。两者的细菌菌落属水平的优势菌属有所不同。
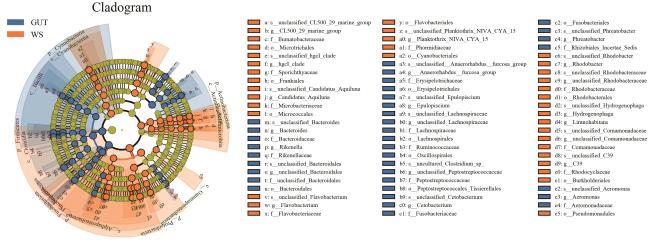
2.3 线性判别
LEfse分析可筛选不同分组间具有统计学差异的生物学标记,检测具有统计学意义的特征值,并将其定位到相应的类群。由图6可知,在门水平,放线菌门、蓝藻菌门等在土壤组中具有重要功能,梭杆菌门、厚壁菌门在水蛭肠道菌群具有重要功能;在属水平,黄杆菌属、氢噬胞菌属和红细菌属(Rhodobacter)等在土壤分组中具有重要功能,理研菌属(Rikenella)和气单胞菌属等在水蛭肠道菌群具有重要功能。
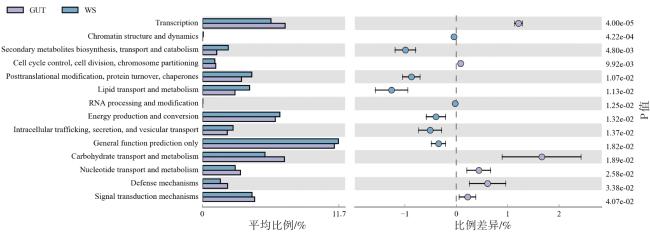
2.4 功能预测
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
3 结论与讨论
肠道微生物对宿主的健康和代谢具有重要作用。测序技术的发展有利于促进对微生物群落的全面了解。肠道微生物的变化是由宿主特性(宿主的饮食、年龄和行为)和外部环境因素引起的。本研究对宽体金线蛭肠道细菌群落进行了鉴定,发现其肠道细菌群落与土壤中的细菌群落有较小的相似性。土壤中的细菌多样性和丰富度明显较高。厚壁菌门、变形杆菌和拟杆菌门是肠道细菌群落的优势菌门,相关研究发现在一些甲壳类动物的肠道中,优势细菌门为厚壁菌门,如黑虎虾[14]、中华绒螯蟹[15]和南美白对虾[16],本研究结果与此具有相似性。变形杆菌具有兼性厌氧或必需厌氧的特点,能够适应一系列的好氧条件,具有更强的营养获取能力[17],因此是肠道优势菌群之一,有助于维持肠道厌氧环境的动态平衡。在所有的肠道样本中均未鉴定出类杆菌门的菌种,或者比例较低。可能是由于其他肠道微生物的生物聚合物具有降解功能。
综上,本研究使用高通量测序技术测定了金线蛭肠道和土壤中的细菌群落。结果表明,该物种肠道和土壤样品中的细菌群落多样性和结构存在明显差异,两者间的优势菌门和菌属存在差异,细菌群落的相似性较小,且在转运通路、碳循环通路和免疫等方面存在差异。表明土壤对肠道细菌群落的建立影响不大。关于土壤和宽体金线蛭肠道细菌群落差异的原因还有待进一步探讨。